Align variables with data dictionary
If using a dataset that is not aligned with the MSF data dictionary, the variable names and values must be standardised. This is because later parts of the template expect certain variable names and values.
This process can take some time and requires looking back-and-forth between your dataset, RStudio, and the MSF data dictionary. In this example, here are the steps taken:
Review the MSF data dictionary
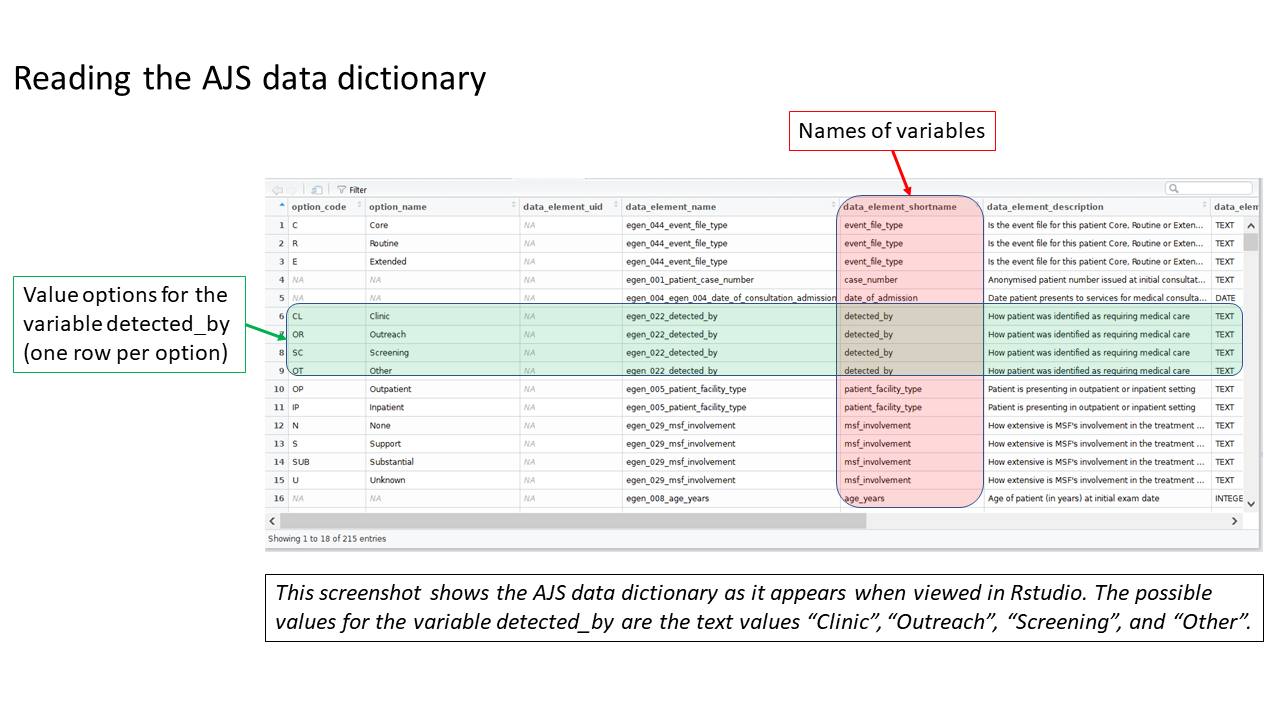
Uncomment and run this command, found in the prep_nonDHIS_data chunk, to view the MSF data dictionary for the disease (“AJS” in this example).
# Creates object linelist_dict using the msf_dict function from the sitrep package
linelist_dict <- msf_dict("AJS", compact = FALSE) %>%
select(option_code, option_name, everything())The dataframe linelist_dict should appear in your Environment pane. You can view the data dictionary by running the command View(linelist_dict) (note capital V), or by clicking on linelist_dict in the Environment pane.
Clean the variable names
These steps standardize how your variable names are written, such as changing spaces and dots to underscores ("_"). Uncomment these lines of code in the prep_nonDHIS_data chunk.
First, make a copy of the dataframe linelist_raw but with a new name: linelist_cleaned. Throughout the template you will modify and improve this linelist_cleaned dataframe. However, you can always return to the linelist_raw version for reference.
## make a copy of your orginal dataset and name it linelist_cleaned
linelist_cleaned <- linelist_rawSecond, use the clean_labels() function from the epitrix package to fix any variable names with non-standard syntax. These code lines take the column names, clean them, and then store the cleaned names in the vector cleaned_colnames. The second code line over-writes the old column names of linelist_cleaned with the improved ones.
If you have a variable named “#” it may be removed by the clean_labels() function. You can protect it by adding the argument protect = "#".
# Store cleaned column names
cleaned_colnames <- epitrix::clean_labels(colnames(linelist_raw))
# Overwrite variable column names with clean ones
colnames(linelist_cleaned) <- cleaned_colnamesMatch variables to align with data dictionary
Standardized variable names are required for this template to work smoothly, and the variable names in our Am Timan dataset do not align with the names this template expects.
We need to change many variable names, and R4Epis developed a special function to help map variables to the expected variable names. The template also offers code that you can uncomment if there are only a few variable names to change.
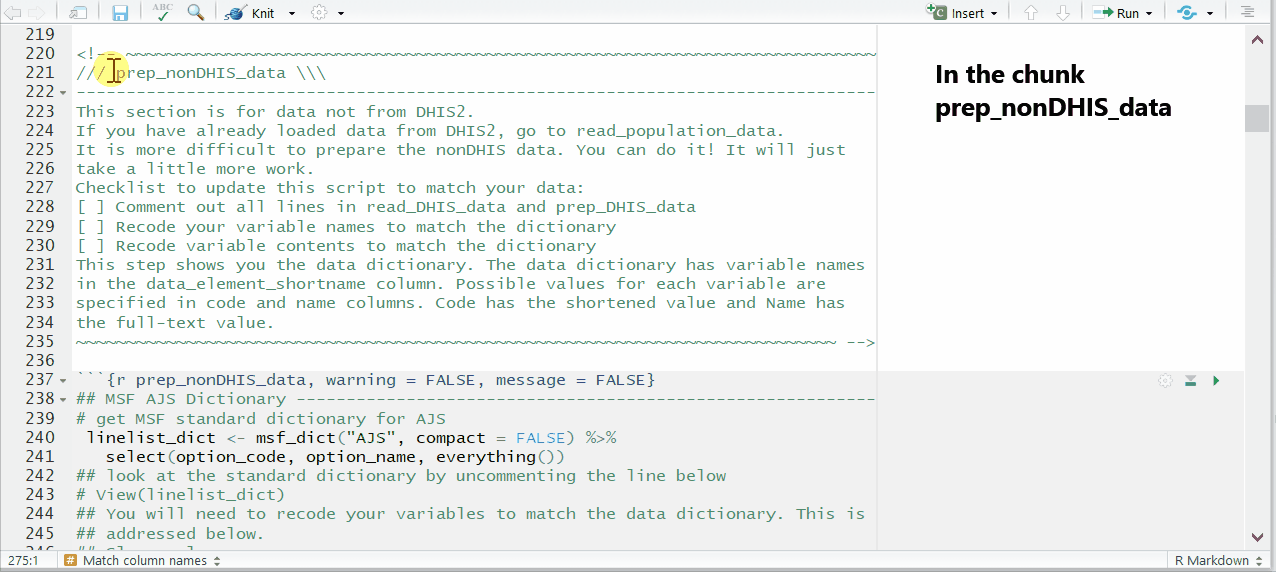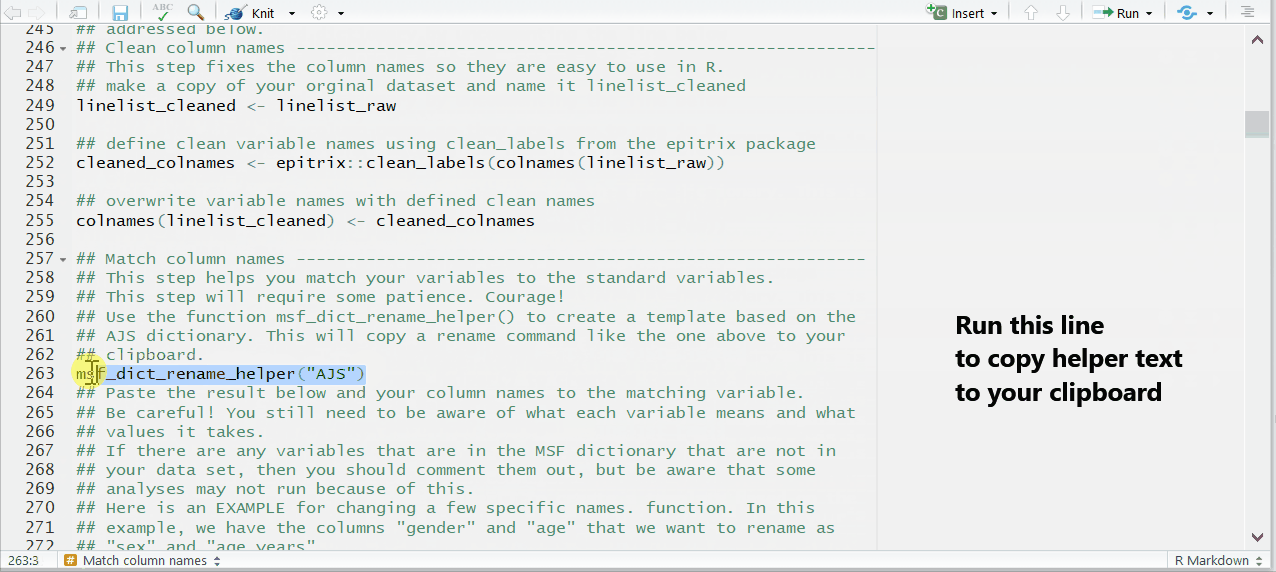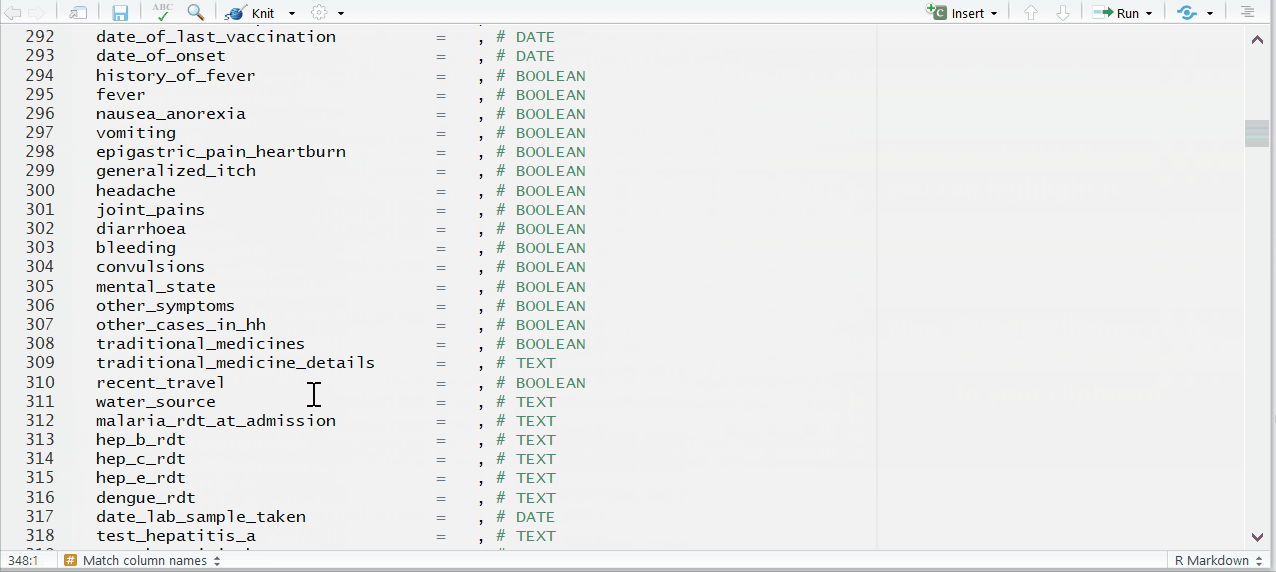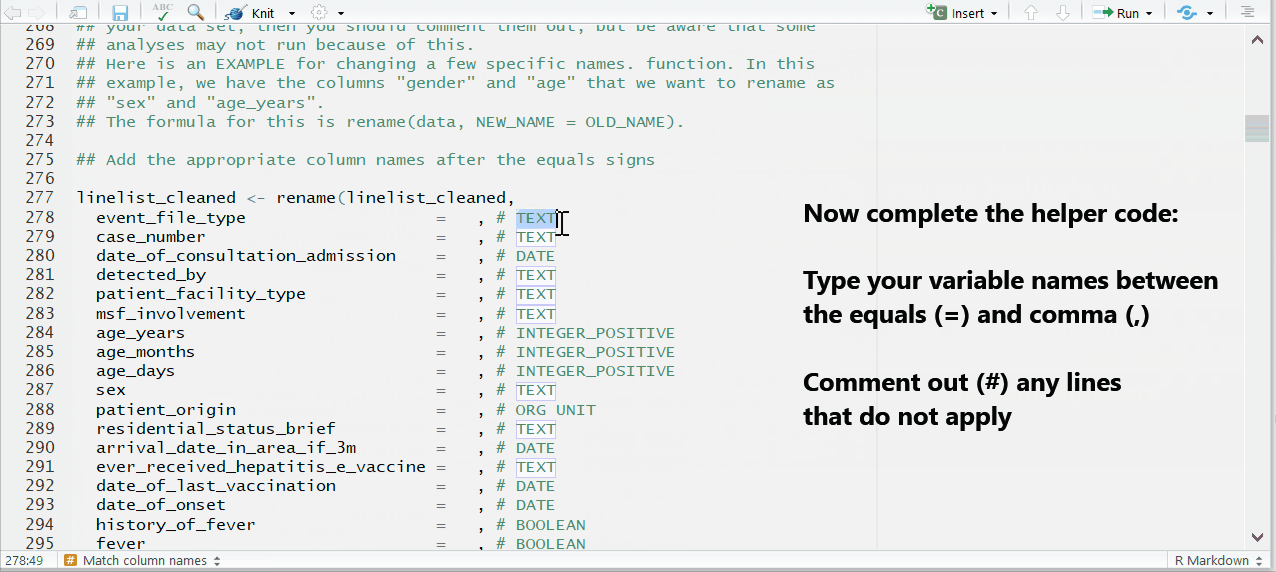
Paste the disease-specific rename helper into the script
This is done by uncommenting and running a command found in the match_column_names section—near the end of the chunk prep_nonDHIS_data. We run the command msf_dict_rename_helper() with “AJS” to get the correct helper for this case study.
msf_dict_rename_helper("AJS")Running this command copies a block of code text to our clipboard. Now, paste from your clipboard into the template in the bottom of the code chunk. For the AJS template, the code that is pasted will look like the code below. This code uses the function rename() to change variable names.
Be sure to paste the AJS rename helper code into an existing R code chunk (e.g. the bottom of prep_nonDHIS_data), not into the white space of the RMarkdown script.
Complete the mapping of variable names
The variables will be in alphabetical order, sorted by if the appear in the template (REQUIRED) or not (optional).
Some variables—such as lab results or symptoms—are listed as REQUIRED, but are not absolutely necessary for the template to run. If you do not have a variable and are unsure if it’s necessary, do a search in the template to see if it’s marked as OPTIONAL in the code chunk.
To the right of each equals sign, and before each comma, type the exact names of the variables from your dataset that correspond to the expected MSF data dictionary variables on the left. The last un-commented line should not have a comma at the end.
Be sure to comment (#, ctrl + shift + c) out the lines of each data dictionary variable in the helper that is not present in your dataset!

If you see this error:
Error in is_symbol(expr) : argument "expr" is missing, with no default
then you likely forgot to comment (#) the line for a variable you did not use.
And now you can see the result:
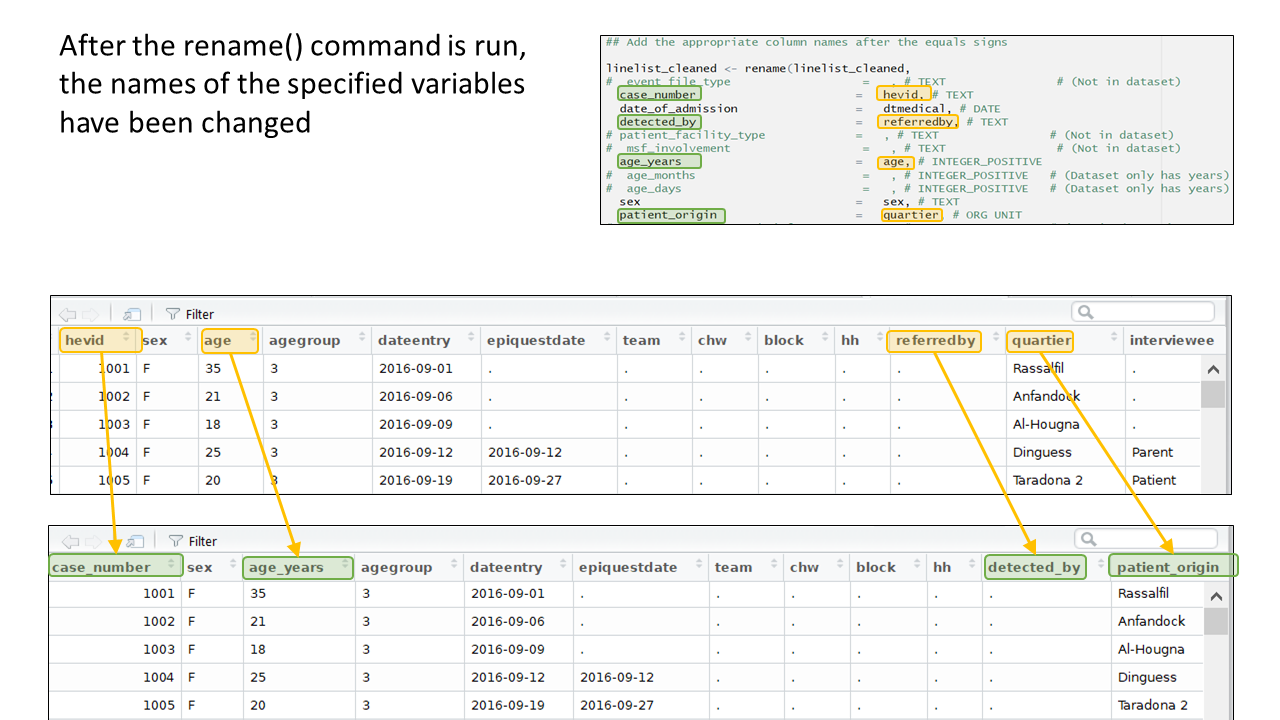
Below is the code for the variable assignments used in this case study walk-through. You can use or reference this when building your template for this case study. You can copy this entire code chunk using the copy icon that appears in the top-right corner.
## Below are the column assignments used in this case study example
linelist_cleaned <- rename(linelist_cleaned,
# age_days = , # INTEGER_POSITIVE (Dataset only has years)
# age_months = , # INTEGER_POSITIVE (Dataset only has years)
age_years = age, # INTEGER_POSITIVE (REQUIRED)
bleeding = medbleeding, # BOOLEAN (REQUIRED)
case_number = hevid, # TEXT (REQUIRED)
chikungunya_onyongnyong = chik, # TEXT (REQUIRED)
# convulsions = , # BOOLEAN (Not in dataset)
date_of_consultation_admission = dtmedical, # DATE (REQUIRED)
# date_of_exit = , # DATE (Not in dataset)
date_of_onset = dtjaundice, # DATE (REQUIRED)
dengue = dengue, # TEXT (REQUIRED)
# dengue_rdt = , # TEXT (Not in dataset)
diarrhoea = meddiar, # BOOLEAN (REQUIRED)
# ebola_marburg = , # TEXT (Not in dataset)
epigastric_pain_heartburn = medepigastric, # BOOLEAN (REQUIRED)
exit_status = outcomehp, # TEXT (REQUIRED)
fever = medfever, # BOOLEAN (REQUIRED)
generalized_itch = meditching, # BOOLEAN (REQUIRED)
headache = medheadache, # BOOLEAN (REQUIRED)
hep_b_rdt = medhepb, # TEXT (REQUIRED)
hep_c_rdt = medhepc, # TEXT (REQUIRED)
hep_e_rdt = medhevrdt, # TEXT (REQUIRED)
# history_of_fever = , # BOOLEAN (Not in dataset)
joint_pains = medarthralgia, # BOOLEAN (REQUIRED)
# lassa_fever = , # TEXT (Not in dataset)
# malaria_blood_film = , # TEXT (Not in dataset)
malaria_rdt_at_admission = medmalrdt, # TEXT (REQUIRED)
mental_state = medmental, # BOOLEAN (REQUIRED)
nausea_anorexia = mednausea, # BOOLEAN (REQUIRED)
other_arthropod_transmitted_virus = arbovpcr, # TEXT (REQUIRED)
other_cases_in_hh = medothhhajs, # BOOLEAN (REQUIRED)
# other_pathogen = , # TEXT (Not in dataset)
other_symptoms = medother, # BOOLEAN (REQUIRED)
patient_facility_type = hospitalised, # TEXT (REQUIRED)
patient_origin = quartier, # ORG UNIT (REQUIRED)
sex = sex, # TEXT (REQUIRED)
test_hepatitis_a = medhavelisa, # TEXT (REQUIRED)
test_hepatitis_b = medhbvelisa, # TEXT (REQUIRED)
test_hepatitis_c = medhcvelisa, # TEXT (REQUIRED)
test_hepatitis_e_genotype = hevgenotype, # TEXT (REQUIRED)
# test_hepatitis_e_igg = , # TEXT (In same variable as elisa result)
test_hepatitis_e_igm = hevrecent, # TEXT (REQUIRED)
test_hepatitis_e_virus = medhevelisa, # TEXT (REQUIRED)
# time_to_death = , # TEXT (Not in dataset)
# typhoid = , # TEXT (Not in dataset)
vomiting = medvomit, # BOOLEAN (REQUIRED)
# water_source = , # TEXT !(Split across many variables)
yellow_fever = yf, # TEXT (REQUIRED)
# arrival_date_in_area_if_3m = , # DATE (optional)
date_lab_sample_taken = medblooddt, # DATE (optional)
# date_of_last_vaccination = , # DATE (optional)
# delivery_event = , # TRUE_ONLY (optional)
detected_by = referredby, # TEXT (optional)
# event_file_type = , # TEXT (optional)
# ever_received_hepatitis_e_vaccine = , # TEXT (optional)
# foetus_alive_at_admission = , # TEXT (optional)
# lab_comments = , # TEXT (optional)
# msf_involvement = , # TEXT (optional)
patient_origin_free_text = block, # TEXT (optional)
pregnancy_outcome_at_exit = medppoutcome, # TEXT (optional)
pregnant = medpreg, # TEXT (optional)
# recent_travel = , # BOOLEAN (optional)
# residential_status_brief = , # TEXT (optional)
# traditional_medicine_details = , # TEXT (optional)
# traditional_medicines = , # BOOLEAN (optional)
treatment_facility_site = hpid, # TEXT (optional)
# treatment_location = , # ORGANISATION_UNIT (optional)
trimester = medpregtri # TEXT (optional)
)